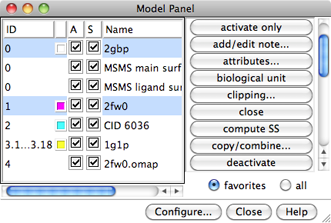
The Model Panel 
The Model Panel lists the models in Chimera and conveniently enables
many operations upon them.
There are several ways to start
the Model Panel, a tool in the General Controls category.
Each file of atomic coordinates opened in Chimera becomes a
model with an associated model ID number.
Surfaces and other types of models also have ID numbers.
Some tools in Chimera create models that are hidden
from the Model Panel, however.
A molecular structure file may contain multiple sets of coordinates
for the same set of atoms. In PDB files,
these coordinate sets are delimited with MODEL
and ENDMDL records.
Appending a
submodel number to the primary model number allows uniquely
specifying atoms despite multiple occurrences of the “same”
atom (same atom name, residue name, and residue number) in the file.
Submodel numbers are assigned sequentially starting with 1
(#0.1, #0.2, etc.).
Multiple orbitals in a Gaussian cube file
are also loaded as submodels.
The term “models” will be used to indicate submodels
and/or models that are not subdivided into submodels
because they are operationally the same:
each can be handled independently of the others and
listed separately in the Model Panel.
At first, the submodels in a model may be collapsed into a single row
(for example, model #3 in the figure below),
but the listing can be expanded to the individual submodels using the
group/ungroup function.
The figure shows columns:
Individual models or multiple models can be
chosen with the left mouse button.
A block of models can be chosen by dragging, or by
clicking on the first (or last) line in the desired block
and then Shift-clicking on its last (or first) line.
Ctrl-click toggles the state (chosen or not) of a single line.
Once one or more models have been chosen
within the left side of the Model Panel, any of several
functions represented by buttons on the
right side of the panel can be executed.
Just the favorites or all functions can be listed, using the
setting below the list. Functions can be added to or removed from the
favorites by showing all functions and using the Fav checkboxes.
The Configure... button allows
customizing which columns appear in the left side of the panel and
which functions are applied to double-clicked models.
Functions:
- activate
- activate for motion;
the opposite of deactivate
- activate all
- activate all models even if not chosen in the left side of the panel
(the previous pattern of activities is saved and can be restored using
the button's alternate function, restore activities)
- activate next
- activate the next applicable (non-skipped) model
in the list
- activate only
- activate the chosen model(s) and deactivate all the others
- activate previous
- activate the previous applicable (non-skipped) model
in the list
- add hydrogens...
add hydrogens to the chosen model(s); equivalent to using
AddH
- add/edit note...
add a text description for the model (the Model Panel can be
configured to show these descriptions
in a Note column; see also
session notes)
- attributes...
list model attributes and allow changes
- biological unit
- add copies to generate the biological unit
according to BIOMT matrices
in the input file; full atomic copies are generated for N-mers of N ≤ 6,
whereas for N-mers of N ≥ 7, the copies are generated as
low-resolution surfaces
from Multiscale Models.
Not all structures include BIOMT matrices.
See also: fetching PDB-biounit
(biological assembly) and PQS files
- clipping...
open the
Per-Model Clipping tool
- close
- close (remove) the model(s)
- color by SS... color peptides/proteins by secondary structure
with the Color
Secondary Structure tool
- compute SS
- define the secondary structure of peptides/proteins in the chosen model(s)
using ksdssp with the specified
parameter values. Hitting return (Enter) or clicking OK runs
the calculation and dismisses the dialog, while clicking Apply
runs the calculation without dismissing the dialog. Clicking Save as
Defaults saves the parameter settings in the
preferences file;
subsequently, those settings will be used with
the ksdssp algorithm
when peptide/protein structures that lack secondary structure assignments are
read in, when
MatchMaker is used with the
Compute secondary structure assignments option,
or when compute SS is specifically invoked.
- copy/combine...
combine multiple molecule models into a single model or make a copy
of a single molecule model
(see also the command combine). The
new model will be assigned the specified ID number and its name
will appear in the Model Panel and other model lists.
A coordinate system (reference model) must be specified.
The reference model's untransformed coordinates (if part of the
combination) and current transformation will be carried over
into the new model. The coordinates of any other model in the
combination will be transformed relative to the reference model.
To ensure that residues in the new model will be uniquely specifiable, it is
necessary to allow either renaming chains (changing chain IDs)
or renumbering residues when the input models contain more than one chain
with the same ID. The original or source model(s) can be closed.
Clicking OK generates the new model and dismisses the dialog,
while clicking Apply generates the new model
without dismissing the dialog. Changes in chain identifiers or residue
numbering are reported in the Reply Log.
- deactivate
- deactivate for motion;
the opposite of activate
- focus
- focus the view on the chosen model(s) and set the
center of rotation method to
center of view (as if using the command
focus on those models)
- group/ungroup
- collapse multiple chosen entries into a group with a specified name,
or expand a single chosen entry that is already a group into its contents;
grouping is hierarchical (groups may contain other groups)
- hide
- disable display of the chosen model(s) (see
display hierarchy);
the opposite of show
- match... open the
MatchMaker tool to superimpose structures with similar sequences
and folds;
at least two molecule models must be chosen for this operation to be available
- rainbow...
color the chosen model(s) over a range with the
Rainbow tool
(see also the command rainbow)
- Ramachandran plot...
show the distribution of peptide φ,ψ angles
(a Ramachandran
plot)
for each chosen model that contains protein; also implemented as the command
ramachandran
- rename...
change the name of each chosen model and/or group.
The name is initially based on input filename or assigned by Chimera when
creating the model (for multi-Mol2 files, see also the
New Molecules preferences).
Another way to change the model name
attribute
is with the command setattr.
- render attribute...
open the
Render/Select by Attribute tool to depict (with color, etc.)
or select atoms, residues, and molecule models
by their attribute values
- save sphgen (available when a
sphere file has been opened)
- save spheres or atoms as a sphere file in the
sphgen format used by
DOCK
- select
- select
the chosen model(s) for subsequent operations or commands
- select chain(s)...
select
subsequently specified chains in the chosen model(s).
The chains are identified in two ways, by ID and by covalent connectivity,
and the listing by ID includes chain descriptions from the PDB, if available.
Any combination of chains from the two identification methods can be
chosen for selection.
- sequence...
show chain sequences (start the
Sequence tool)
- show
- enable display of the chosen model(s) (see
display hierarchy);
the opposite of hide
- show all atoms
- show the chosen model(s) and display all atoms within them
- show next
- show the next applicable (non-skipped) model
in the list
- show only
- show the chosen model(s) and hide all of the others
- show previous
- show the previous applicable (non-skipped) model
in the list
- surface main
- for the chosen model(s), display the molecular surface of any atoms
categorized as main
- tile...
open Tile
Structures
to arrange the (two or more) chosen models in a plane
- toggle active
- toggle the status of each chosen model between
activated and deactivated for motion
(make the status of each its current opposite)
- trace backbones
- in each chosen model,
display only a simplified trace for bonded chains containing
amino acid or nucleic acid residues, consisting of real backbone bonds
(-[N-CA-C]n- in
peptides and -[O5'-C5'-C4'-C3'-O3'-P]n- in nucleic acids)
- trace chains
- in each chosen model,
display only a simplified trace for bonded chains containing
amino acid or nucleic acid residues, consisting of one atom per residue
(connect the CA atoms in peptides, C4' atoms in nucleic acids)
- transform as...
transform each (initially) chosen model using the rotation/translation matrix
of the single model chosen when the dialog's OK button is clicked
- UniProt info...
start PDB/UniProt
Info to retrieve
UniProt
annotations and map them to sequence
- write PDB
- save models
as PDB files
Close dismisses the Model Panel. Help
opens this manual page in a browser window.
Model Panel Configuration
The Configure... button brings up another panel with three
sections for customizing the Model Panel:
-
The Buttons section
was used in older versions of Chimera to classify
functions as frequently or infrequently used.
Currently Chimera uses a favorites list instead, where just favorites
or all functions can be shown in the Model Panel.
To configure which functions are favorites,
change from showing just favorites to showing all
in the Model Panel, then use the Fav checkboxes.
-
The Columns section
determines which model attributes will
be shown in the left side of the Model Panel.
The descriptions below are in the order in which the columns
would be shown, from left to right.
- ID - model (and submodel, if applicable) number
- [color] - for each molecule model,
a color well
showing the model-level color;
blank for molecular surface models and
other model types.
Clicking a color well
opens the Color Editor and
allows the color to be changed.
- Active - whether the model is
activated for motion;
see activate
and deactivate
- Shown - whether the model is shown;
see hide and show
- Name - model name; see rename
- Skip
- whether to skip the model (leave it unchanged) when using
next/previous functions.
These functions allow easily switching which single model is shown and/or
active, based on main model number (submodels are not treated separately).
- Note - user-added descriptive text for the model; see
add/edit note
- Input file - for local files, a pathname (relative if the file
is in a subdirectory of the working directory, otherwise full); for files
fetched from databases,
an identifier or filename for that database entry
Show model color behind model name shows the
model-level color of each molecule model
in the Name column, with the text of the name when the model is
chosen, otherwise with a swatch of color behind the name.
- The Double Click section
controls which functions will be executed upon
a model when the model is double-clicked in the left side of the Model
Panel. The default is attributes;
however, there can be zero or many functions in the list for execution.
Clicking an entry in the Function menu
on the right causes it to appear in the Execution list on the left.
Clicking an entry in the Execution list removes it from that list.
The functions will be executed in the order listed.
Model Panel customization settings are stored in a user's
preferences file.
UCSF Computer Graphics Laboratory / May 2022