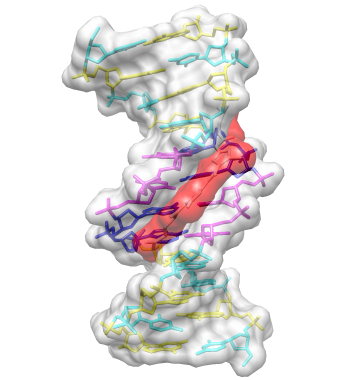
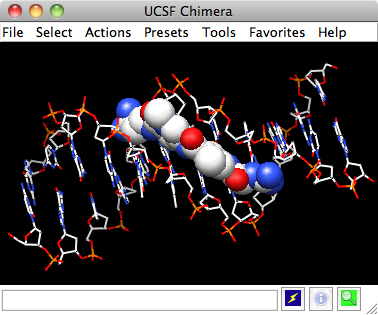
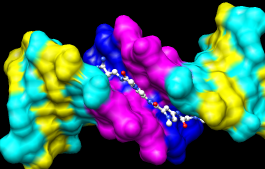
DNA helix with bound netropsin
DNA helix with bound netropsin
This tutorial provides an overview of basic features in Chimera. You can interact with Chimera using menus and/or commands. The basic features of Chimera are available either way, but not all command functions are available in menus or graphical interfaces, and not all menu or graphical interface functions are available in commands. Thus, it is useful to become familiar with both ways of interacting with Chimera.
The Working with menus and Working with commands sections are independent of each other and (for the most part) cover identical operations, accomplished in different ways. If you go through both sections, you can skip portions that cover issues you already understand. You can also go back and forth between the sections to see the correspondence between menu and command operations.
| Item | Example | Description |
| Keyboard key | Ctrl | The control key |
| Mouse key | Btn1 | Mouse button 1 (left button) |
| Menu action | File→Open | File Menu bar pulldown, followed by Open |
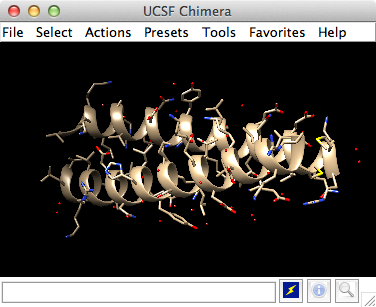
UCSF Chimera with 1zik
Start Chimera by clicking or doubleclicking the Chimera icon
![]() (depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its installation location.
(depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its installation location.
A splash screen will appear, to be replaced in a few seconds by the main Chimera window containing either the graphics display or the Rapid Access list of recently used files (it doesn't matter which, since opening a structure will automatically switch the display to the graphics window). If you like, enlarge the window by clicking and dragging its lower right corner. The window can also be moved by clicking its top bar and dragging.
Now open a structure. Choose File→Fetch by ID from the menu and enter 1zik as the ID of the structure to fetch from the PDB. The structure will appear in the graphics window; it is a leucine zipper formed by two peptides.
The default initial display is ribbons. To also display atoms:
Actions→Atoms/Bonds→showThis shows all of the atoms and bonds in the structure, except that those in the peptide backbone are suppressed by the ribbon display. How to indicate specific parts of a structure for display, coloring, etc. is discussed below. Initially, heteroatoms (atoms other than carbon) are color-coded by element: oxygens red, nitrogens blue, etc. The carbons retain the model color, in this case tan.
Hide ribbons to reveal the backbone atoms, then show ribbons again:
Actions→Ribbon→hide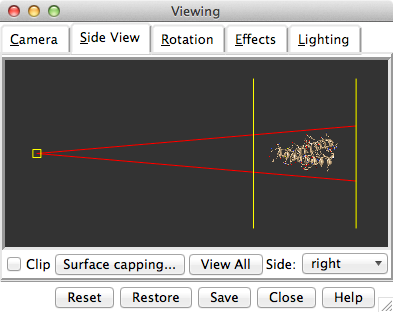
Side View showing 1zik
Show the Side View for interactive scaling and clipping (menu: Tools→Viewing Controls→Side View). By default, the Side View is also listed in the Favorites menu.
It shows a tiny version of the structure. Try moving the eye position (the small square) and the clipping planes (vertical lines) by clicking and dragging with the left mouse button. The Side View will renormalize itself after movements, so that the eye or clipping plane positions may appear to “bounce back,” but your adjustments have been applied.
| Mouse button | Modifier | Action |
| Btn1 (left button) | |
Rotation |
| Btn2 (middle button) | |
XY Translation |
| Btn3 (right button) | |
Scaling |
| Btn1 | Ctrl | Picking (selection) |
| Btn1 | Ctrl-Shift | Addition to (removal from) selection |
Try manipulating the structure in the main window with the mouse. By default:
When the mouse focus is in the graphics window (you may need to click into it if you have been interacting with a different window), hovering the mouse cursor over an atom or bond without clicking any buttons will show identifying information in a pop-up “balloon.” The balloon will disappear when the cursor is moved away. For an atom, the balloon information is of the form:
res-name res-num.chain atom-nameYou can see from the balloons that this structure contains two peptide chains, A and B, and water (HOH residues), also with chain identifiers A and B.
In combination with modifier keys,
the mouse buttons have additional functions. By default,
picking from the screen (a type of selection)
is done by Ctrl-clicking an atom or bond
with the left mouse button, Btn1.
You can also drag out a selection area with
Ctrl-Btn1
(sweep out an area before releasing).
Shift-Ctrl-Btn1
adds to or toggles an existing selection.
The selection is outlined in green, and placing the mouse cursor over
the green magnifying glass icon  near the bottom right corner of the window reports what is
selected in a pop-up “balloon.”
near the bottom right corner of the window reports what is
selected in a pop-up “balloon.”
The arrow keys can be used to broaden (↑), narrow (↓), or invert (→) a selection. The hierarchy for broadening and narrowing a selection could include (depending on the initial selection): atom/bond, residue, protein secondary structure element, bonded set of atoms, all atoms with the same chain ID, entire model. When a selection is inverted, the selected atoms become deselected and vice versa.
Spend some time selecting various parts of the structure. An easy way to clear the selection (deselect everything) is to use Ctrl-Btn1 in any blank space in the graphics window.
| Menu Item | Description |
| Atoms/Bonds | Controls the display and representation of atoms and bonds. |
| Ribbon | Controls the display and representation of ribbons. |
| Surface | Controls the display and representation of molecular surfaces. |
| Color | Colors selected objects. Color target can be limited to object types in the all options dialog. |
| Label | Labels selected atoms. The residue submenu labels residues containing the selected atoms. |
| Focus | Focuses the view on the selected atom(s), zooming and translating if necessary. |
| Set Pivot | Sets the center of rotation based on the selected atom(s) without adjusting the view. |
| Inspect | Launches the Selection Inspector;
same as clicking . |
| Write List | Writes a list of the currently selected objects to a parsable text file. |
| Write PDB | Writes the coordinates of the currently selected atoms to a PDB file. |
In general, operations performed with the Chimera Actions menu apply to the current selection. Selections can be made in many ways, including with the Select menu or with the mouse (as described above). When nothing is selected, the Actions menu applies to everything.
The following will color all residues named LYS hot pink.
Select→Residue→LYS
The selection is highlighted in green,
and the magnifying glass icon
near the bottom right corner of the window is also green,
indicating that something is selected.
Clearing the selection (deselecting) beforehand will color everything:
Select menu choices also include chain ID, element, and many other categories of atoms and residues. (More complicated selections can be built up by intersecting some of these choices, as shown near the end of Part 2.)
Select→Chain→BOne way to select specific residues or ranges of residues is in the Sequence tool (menu Favorites→Sequence, show sequence for chain A). When the sequence window has mouse focus, placing the cursor over a residue symbol in the sequence shows information for the corresponding structure residue at the bottom of the window. Click-drag a box within the sequence window to select one or more residues (as opposed to simply clicking within the light yellow box, which will select the entire helix), then hide their atoms:
Actions→Atoms/Bonds→hideQuit from the sequence window and display all protein atoms again:
Select→Structure→proteinColoring can be limited to only certain representations, such as atoms only (not ribbons, surfaces, etc.):
Select→Residue→GLUIn the resulting Color Actions dialog:
Coloring by heteroatom is useful for showing functional groups, yet keeping different models distinguishable by their different carbon colors.
Try picking two atoms in different residues (Ctrl-click the first, Shift-Ctrl-click the second). Show residue labels for the atoms you have selected:
Actions→Label→residue→name + specifier(Actions→Label→name would show the atom names instead.) These 3D labels move along with structures and are mainly for interactive use. For figures and movies, 2D Labels are recommended instead.
Promote the selection to the entire residues with the keyboard up arrow ↑ or the following:
Select→BroadenShow only the selected atoms:
Actions→Atoms/Bonds→show onlyClear the selection by Ctrl-clicking in empty space, as if picking “nothing.”
Turn off residue labels, hide ribbon, display all atoms, and color by element:
Actions→Label→residue→offThe by element coloring is the same as by heteroatom except it also color-codes carbons (gray).
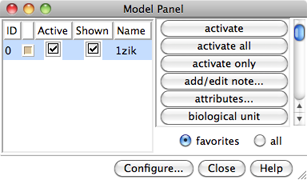
Chimera Model Panel
Generally, each file of coordinates opened in Chimera becomes a model with an associated model ID number. Models are assigned successive numbers starting with 0. The Model Panel lists the current models and enables many operations upon them. Open this tool with Tools→General Controls→Model Panel.
The columns on the left side of the Model Panel show:
Go on to Part 2 below, or exit from Chimera with File→Quit.
Chimera showing netropsin as spheres
With Chimera started as described at the beginning of Part 1, open a different structure. Choose File→Fetch by ID from the menu and enter 1d86 as the ID of the structure to fetch from the PDB. The structure contains the molecule netropsin bound to double-helical DNA, initially shown with ribbons and stylized representations of the nucleic acid sugars and bases.
Rotate, translate, and scale the structure as needed to get a better look (see Using the mouse to review how this is done). Continue moving and scaling the structure as desired throughout the tutorial.
A preset is a predefined combination of display settings. Use the “all atoms” preset, which will show the DNA as wire and netropsin as spheres:
Presets→Interactive 2 (all atoms)Color carbons white, then undisplay the water:
Select→Chemistry→element→C
Remember that hiding atoms does not deselect them; they remain selected,
as indicated by the green magnifying glass icon
near the bottom right of the window,
until the selection is cleared or replaced with a new selection.
Residue names can be identified by looking in the Select→Residue menu or by hovering the cursor over an atom or bond to see information in a pop-up “balloon.” Color the different nucleotides different colors, for example:
Select→Residue→DAAnalogously, color DC residues cyan, DG residues yellow, and DT residues magenta. Clear the selection with Select→Clear Selection or Ctrl-click in empty space.
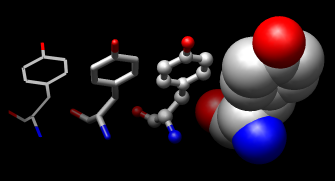
Atoms/Bonds: wire, stick, ball & stick, and sphere
Next, try some different display styles, or representations.
Actions→Atoms/Bonds→sphereShowing ribbon automatically hides the mainchain (backbone) atoms.
Actions→Ribbon→show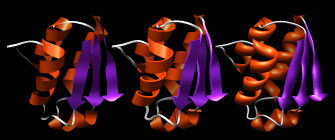
Ribbon: flat, edged, and rounded
DNA can be shown with special nucleotide objects. We will show “lollipops,” boxes, and a ladder.
Actions→Atoms/Bonds→nucleotide objects→settingsIn the resulting Nucleotides dialog:
Show the sequence of chain A and select one or a few residues in the sequence window with the mouse; this selects the corresponding part of the structure. Quit from the sequence window. In the Nucleotides dialog (also under Tools→Depiction in the menu):
Clear the selection (Select→Clear Selection), then use Nucleotides to show the DNA as a ladder:
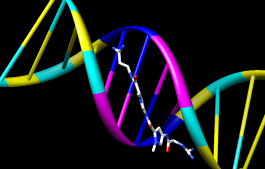
Ribbons and nucleotide ladder
Hide the ribbons and show everything as ball-and-stick:
Actions→Ribbon→hide
Molecular surface (main)
Finally, have some fun with molecular surfaces. There are built-in categories within structures such as main and ligand; when nothing is selected, Actions→Surface→show displays the surface of main.
Actions→Surface→showSurface color can be specified separately from the colors of the underlying atoms. The ligand surface is tan and white because the original model color (tan) is used for surfaces of atoms not explicitly recolored by the user, and above, only the carbon atoms were changed to white. With the ligand still selected, choose Actions→Color→all options... to open the Color Actions dialog. In that dialog:
Clear the selection, change back to a solid surface, and then undisplay the surface.
Select→Clear SelectionAs an example of a more complicated selection process, show the surface of the adenine and thymine deoxynucleotides in chain B only:
The command-line (Tools→General Controls→Command Line) equivalent is much more concise, but requires some knowledge of the atom specification syntax:
Command: surf :da.b,dt.bSometimes it is helpful to make a surface transparent: Actions→Surface→transparency→50%
Choose File→Quit from the menu to terminate the Chimera session.
DNA helix with bound netropsin
Chimera with Command Line
Start Chimera by clicking or doubleclicking the Chimera icon
![]() (depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its installation location.
(depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its installation location.
A splash screen will appear, to be replaced in a few seconds by the main Chimera window containing either the graphics display or the Rapid Access list of recently used files (it doesn't matter which, since opening a structure will automatically switch the display to the graphics window). If you like, enlarge the window by clicking and dragging its lower right corner. The window can also be moved by clicking its top bar and dragging.
Show the Command Line with Tools→General Controls→Command Line. By default, the Command Line is also listed in the Favorites menu.
Now open a structure. To fetch the structure of entry 1zik from the Protein Data Bank (PDB), use the command:
Command: open 1zikThe structure will appear in the graphics window; it is a leucine zipper formed by two peptides.
The default initial display is ribbons. To also display atoms:
Command: display
This shows all of the atoms and bonds in the structure, except that those in the peptide backbone are suppressed by the ribbon display. How to indicate specific parts of a structure for display, coloring, etc. is discussed below. Initially, heteroatoms (atoms other than carbon) are color-coded by element: oxygens red, nitrogens blue, etc. The carbons retain the model color, in this case tan.
Hide ribbons to reveal the backbone atoms, then show ribbons again:
Command: ~ribbon
Command: ribbon
Many commands have “~” versions that perform the opposite function.
Side View showing 1zik
Show the Side View for interactive scaling and clipping:
Command: start Side ViewBy default, the Side View can also be started from the Favorites menu.
It shows a tiny version of the structure. Try moving the eye position (the small square) and the clipping planes (vertical lines) by clicking and dragging with the left mouse button. The Side View will renormalize itself after movements, so that the eye or clipping plane positions may appear to “bounce back,” but your adjustments have been applied.
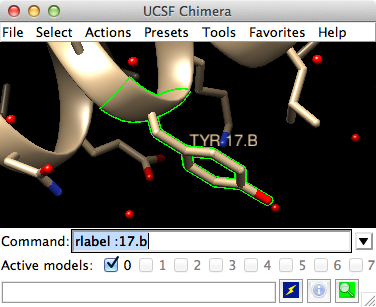
1zik with tyrosine 17 (B chain) selected
Try manipulating the structure in the main window with the mouse. By default:
When the mouse focus is in the graphics window (you may need to click into it if you have been interacting with a different window), hovering the mouse cursor over an atom or bond without clicking any buttons will show identifying information in a pop-up “balloon.” The balloon will disappear when the cursor is moved away. For an atom, the balloon information is of the form:
res-name res-num.chain atom-nameYou can see from the balloons that this structure contains two peptide chains, A and B, and water (HOH residues), also with chain identifiers A and B.
In combination with modifier keys,
the mouse buttons have additional functions. By default,
picking from the screen (a type of selection)
is done by Ctrl-clicking an atom or bond
with the left mouse button, Btn1.
Additionally pressing Shift
adds to or toggles an existing selection.
The selection is outlined in green, and placing the mouse cursor over
the green magnifying glass icon
near the bottom right corner of the window reports what is
selected in a pop-up “balloon.”
The arrow keys can be used to broaden (↑), narrow (↓), or invert (→) a selection. The hierarchy for broadening and narrowing a selection could include (depending on the initial selection): atom/bond, residue, protein secondary structure element, bonded set of atoms, all atoms with the same chain ID, entire model. When a selection is inverted, the selected atoms become deselected and vice versa.
Spend some time selecting various parts of the structure. An easy way to clear the selection (deselect everything) is to use Ctrl-Btn1 in any blank space in the graphics window.
| Symbol | Function | Usage |
| # | model number | # model (integer) |
| : | residue | : residue (name or number) |
| :. | chain ID | :.chain |
| @ | atom name | @atom |
| * | whole wildcard | matches whole atom or residue names, e.g., :*@CA specifies the α-carbons of all residues |
| = | partial wildcard | matches partial atom or residue names, e.g., @C= specifies all atoms with names beginning with C |
| ? | single-character wildcard | used for atom and residue names only, e.g., :G?? specifies all residues with three-letter names beginning with G |
| z< | zone specifier | z<zone or zr<zone specifies all residues within zone angstroms of the indicated atoms, and za<zone specifies all atoms (rather than entire residues) within zone angstroms of the indicated atoms. Using > instead of < gives the complement. |
| & | intersection | intersection of specified sets |
| | | union | union of specified sets |
| ~ | negation | negation of specified set (when space-delimited) |
A Chimera command may include arguments and a target (or atom specification). For example, in the following color command,
Command: color hot pink :lyshot pink is an argument that specifies a color name, and :lys specifies that the target is all residues named LYS.
If no target is specified, the command acts on all applicable items. For example, the following makes everything hot pink:
Command: color hot pinkA basic specification may contain residue names, residue numbers, chain identifiers, and/or atom names (see the table of symbols, right). Note also that commands can be truncated to unique strings.
Command: color yellow :20-22
Command: col gray :20-22.a
Command: ~disp :hoh.a
Command: col cyan :.b
Command: ~ribbon :.b
Command: ~disp :5-10.a,15-20.b
Command: represent sphere @cd2
Command: rep stick
Command: col red :leu.b@ca
The Chimera Quick Reference Guide lists all of the commands and gives some examples of atom specification. It can be accessed by choosing Help→Tutorials from the Chimera menu and clicking the “Chimera Quick Reference Guide” link.
Many other types of specifications can be used, including element symbols and built-in classifications such as solvent:
Command: ~disp solventThe command help can be used to show the manual page for any command:
Command: color blue S
Command: disp protein
Command: help colorAs explained in the manual page, the color command also allows coloring only certain representations. For example, “,a” in the following means atoms only (not ribbons, surfaces, etc.):
Command: col gold,a :glu,lysRestore the original coloring:
Command: col tan
Command: col byhet
Coloring by heteroatom is useful for showing functional groups, yet keeping different models distinguishable by their different carbon colors.
Try picking two atoms in different residues (Ctrl-click the first, Shift-Ctrl-click the second). Unlike the Actions menu, commands do not automatically act on the current selection. However, the current selection can be specified as the target of a command with the word selected, sel, or picked. Show residue labels for the atoms you have selected:
Command: rlabel sel
(The label command shows atom information instead.) The 3D labels from rlabel and label move along with structures and are mainly for interactive use. For figures and movies, 2D Labels are recommended instead.
The following command can be used to promote the selection to the entire residues:
Command: select up(The keyboard up arrow ↑ also broadens a selection, but you may need to click into the graphics window first to use that approach.) Show only the selected atoms:
Command: show selClear the selection by Ctrl-clicking in empty space, as if picking “nothing.”
Turn off residue labels, hide ribbon, display all atoms, and color by element:
Command: ~rlabColoring byelement is the same as byhet except it also color-codes carbons (gray).
Command: ~ribbon
Command: disp
Command: col byelement
Generally, each file of coordinates opened in Chimera becomes a model with an associated model ID number. Models are assigned successive numbers starting with 0.
The Active models line right under the Command Line shows which models are activated for motion. Unchecking the box for 0 makes it impossible to rotate or translate model 0 interactively. Checking the box again restores the movable state. A similar row of checkboxes for toggling model display can be shown using the preferences (menu: Favorites→Preferences, category Command Line).
Remove the model:
Command: close 0Go on to Part 2 below, OR exit from Chimera with the following command:
Command: stop
Chimera with Command Line
With Chimera started and the Command Line opened as described at the beginning of Part 1, fetch the structure of entry 1d86 from the Protein Data Bank (PDB):
Command: open 1d86The structure contains the molecule netropsin bound to double-helical DNA, initially shown with ribbons and stylized representations of the nucleic acid sugars and bases.
Rotate, translate, and scale the structure as needed to get a better look (see Using the mouse to review how this is done). Continue moving and scaling the structure as desired throughout the tutorial.
A preset is a predefined combination of display settings. Use the “all atoms” preset, which will show the DNA as wire and netropsin as spheres:
Command: preset apply int 2Color carbons white, then undisplay the water:
Command: color white C
Command: ~disp solvent
Residue names can be identified by looking in the Select→Residue menu or by hovering the cursor over an atom or bond to see information in a pop-up “balloon.” Color the different nucleotides different colors, specifying them by residue name:
Command: color blue :da
Command: color cyan :dc
Command: color yellow :dg
Command: color magenta :dt
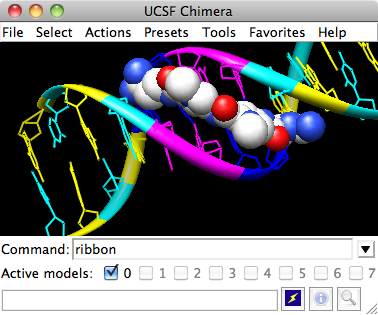
Ribbons and nucleotide ladder
Next, try some different display styles, or representations.
Command: represent sphereNotice that commands but not necessarily their keyword arguments can be truncated to unique strings. For example, the command represent can be shortened to repr or rep but not re (because other commands also start with re), whereas its keywords stick, sphere, etc. cannot be truncated. If the truncation is not unique, one of the corresponding commands will be executed, but it may not be the one intended.
Command: repr bs :.a
Command: rep stick
Showing ribbon automatically hides the mainchain (backbone) atoms.
Command: ribbonDNA can be shown with special nucleotide objects. We will show “lollipops,” boxes with orientation bumps, and then a ladder. You can copy and paste into the Command Line. The command-line contents can be edited, and past commands can be accessed using the up and down arrow keys or Ctrl-p (previous) and Ctrl-n (next).
Command: ribrep edged
Command: ribr rounded
Command: nuc side tube/slab shape ellipsoid orient false style skinnyTo return to more general display styles, turn off the nucleotide objects:
Command: nuc side tube/slab shape box orient true style skinny :8-10.a
Command: nuc side ladder radius 0.3
Command: ~nucHide the ribbons and show everything as ball-and-stick:
Command: ~ribbon
Command: rep bs
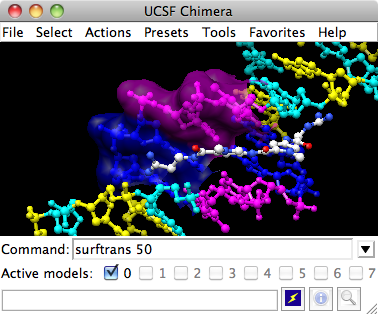
Chimera showing a transparent surface
Finally, have some fun with molecular surfaces. There are built-in categories within structures such as main and ligand; when nothing is specified, surface shows the surface of main.
Command: surfaceSurface color can be specified separately from the colors of the underlying atoms. The ligand surface is tan and white because the original model color (tan) is used for surfaces of atoms not explicitly recolored by the user, and above, only the carbon atoms were changed to white. Show the ligand surface as red mesh:
Command: ~surf
Command: surf ligand
-OR- (equivalent)
Command: surf :nt
Command: surfrep meshParts of a surface can be shown:
Command: color red,s ligand
Command: surfrep solid
Command: ~surfSometimes it is helpful to make a surface transparent:
Command: surf :da,dt
Command: ~surf
Command: surf :da.b,dt.b
Command: transp 50,sWhen finished, exit from Chimera:
Command: stop now
DNA helix with bound netropsin